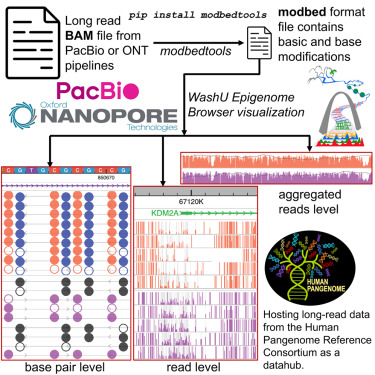
In recent years, third-generation long-read sequencing technologies such as Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) have made significant advancements in read length, throughput, yield, and analytical methods. These technologies directly sequence single molecules of nucleic acids in real-time, generating longer reads than second-generation sequencing platforms. Long-read sequencing has had a profound impact on all major areas of genomics. For instance, longer reads contribute to improved genome assembly. The Human Pangenome Reference Consortium now commonly employs long-read sequencing to construct individual genomes of reference quality, better representing human diversity.
In addition to generating longer reads, single-molecule-based sequencing can also detect the chemical modifications of bases. For example, PacBio can detect 5-methylcytosine (5mC) modifications by analyzing the dynamic features composed of pulse width and pulse interval duration. ONT sequencing quantifies readings of electrolytic signals sensitive to base modifications, making it suitable for detecting chemical modifications such as 5mC. Researchers have leveraged these new capabilities to invent various techniques that convert chromatin biological signals into base modifications. For instance, Fiber-seq utilizes N6-adenine (6mA) marking to study regulatory DNA and nucleosome positioning at single-chromatin fiber resolution. There are tools available for visualizing such data, including desktop software like IGV, command-line tools like modbamtools, and web-based genome browsers like JBrowse2. They typically display individual sequencing reads in a heatmap, where each row represents the chemical modification of a read, using red/blue colors in columns to indicate methylation status. However, these displays often lack statistical information such as methylation percentage and base modification data for each long read.
To address this limitation, the WashU Epigenome Browser has introduced the Modbed track data type. This data track type allows for the visualization of modification details at the level of individual long reads, as well as the display of merged modification information for one or more long reads within a dynamic resolution range. Modbed files can be uploaded as local files for visualization or submitted for visualization on the WashU Epigenome Browser through accessible URLs. The WashU Epigenome Browser can be accessed via: https://epigenomegateway.wustl.edu/. This new update is published in Cell Genomics
The Modbed track primarily offers three types of visualizations based on the size of the genomic interval the user is browsing. If you are examining a region of a few tens of base pairs, the modification information for each long read will be displayed as shown in the following figure:
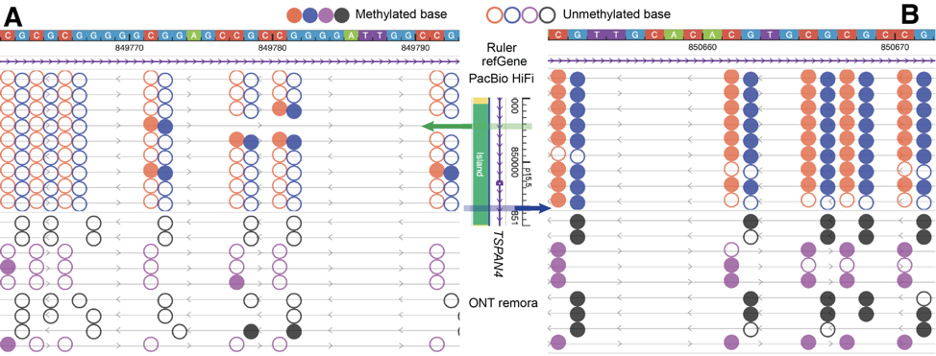
Upon further zooming into this region, the browser will merge the modification information of adjacent CpG sites on each long read, as shown in Part A of the following figure. In Part A, each row still represents a long-read sequence, allowing for easy inspection of the modification information for each long read. Each bar represents the merged methylation level of adjacent CpG sites, ranging from 0 (completely unmethylated) to 1 (completely methylated). In the same region, by right-clicking on the track, you can switch to a heatmap mode, transforming the display into Part B of the figure. In this classic heatmap, red or purple represents the methylation level, blue represents the unmethylated level, and horizontal gray lines indicate the sequencing depth of the long-read sequences.
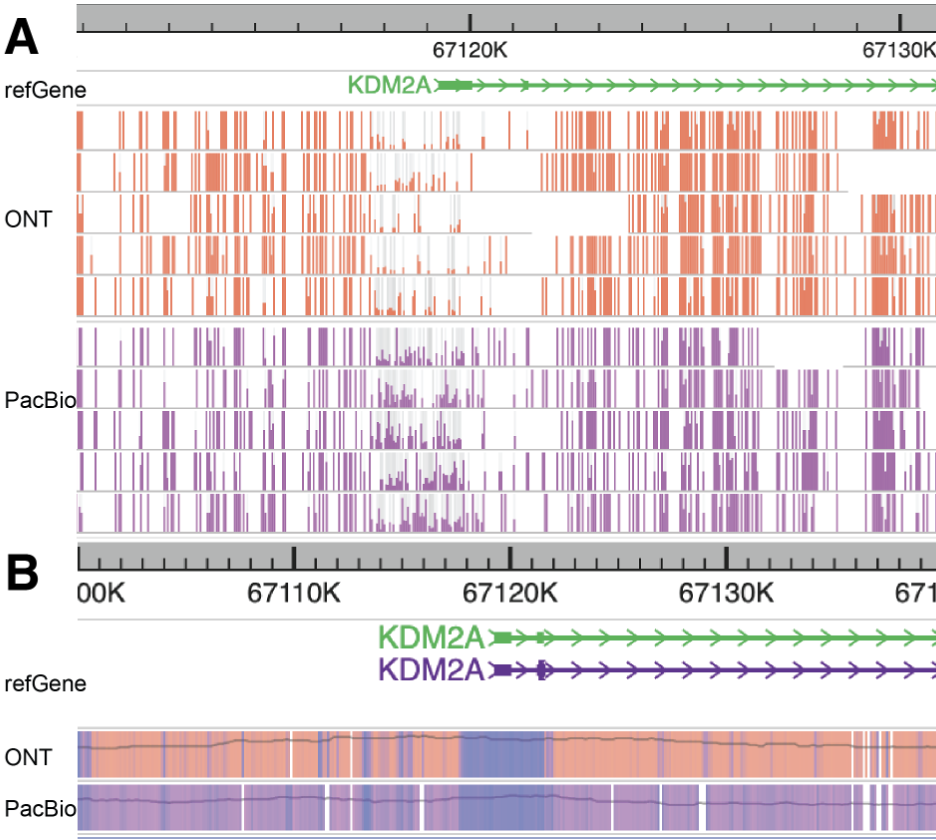
If a user continues to zoom in on this region, the browser will not only merge adjacent CpG sites on each long read but also aggregate all the long reads in this region to display a comprehensive modification/methylation profile, as shown in the figure below.
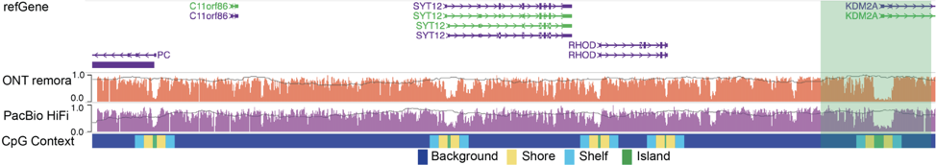
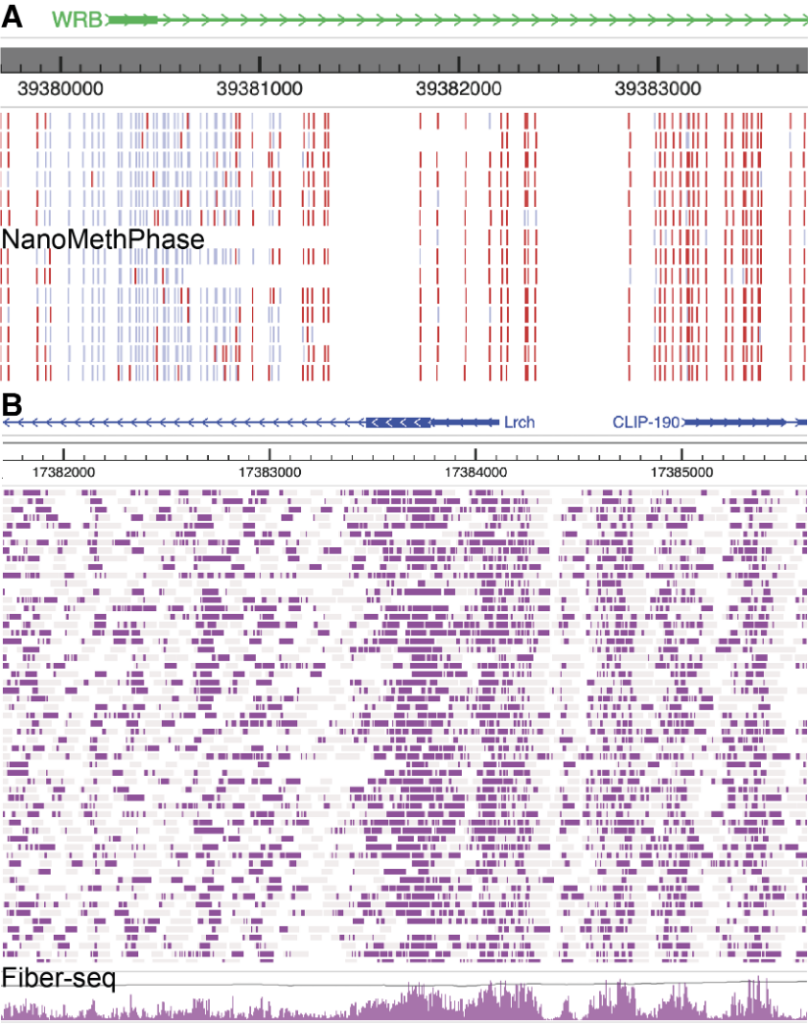
A user can also use the modbed track to display other sequencing data based on third-generation sequencing technologies, such as NanoMethPhase and Fiber-seq, as shown in the figure below:
The development of modbed track in the WashU Epigenome Browser is led by Dr. Daofeng Li, Assistant Professor in the Department of Genetics, Washington University School of Medicine. The project was initiated by the Wang Lab around December 2022 in collaboration with Dr. John A. Stamatoyannopoulos, Director of Altius Institute for Biomedical Sciences.
What is the WashU Epigenome Browser?
WashU Epigenome Browser is an online platform developed by the Wang Lab to help researchers visualize epigenomic data. The browser supports genomic annotation features, numerical data, and chromatin interactions. It is lightweight, customizable, and user-friendly. Users can use it to visualize data locally as well as on the cloud.
Selected publications related to the WashU Epigenome Browser:
Featured in this article:
Daofeng Li, PhD

Daofeng Li is an Assistant Professor of Genetics at Washington University School of Medicine in St. Louis. A member of the Wang Lab, he joined the project of WashU Epigenome Browser in 2011.
Wang Lab

The Wang Lab’s research focuses on understanding the evolution and adaptation of human regulatory networks, particularly their impact on human health and disease. Utilizing integrative and systems approaches, the lab develops statistical and computational algorithms for exploring the human genome, integrating cross-species comparative and high-throughput genomics data. The lab is also involved in developing genome browsers and tools for analyzing high-throughput genomics data, including next-gen sequencing data.